Fibrosis quística
De Wikipedia, la enciclopedia libre
La fibrosis quística (abreviatura FQ), también conocida como mucoviscidosis (del lat. muccus, "moco", y viscōsus, "pegajoso"), es una enfermedad hereditaria frecuente que afecta al organismo en forma generalizada, causando discapacidad progresiva y muerte prematura. La dificultad para respirar es el síntoma más común, emergente de infecciones pulmonares crónicas, las cuales pueden mostrarse resistentes al tratamiento con antibióticos y otros fármacos. La FQ es un trastorno multisistémico que causa la formación y acumulación de un moco espeso y pegajoso, afectando fundamentalmente a pulmones, intestinos, páncreas e hígado. Asimismo, se caracteriza por la presencia de una alta concentración de sal (NaCl) en el sudor, lo que sentó las bases de la prueba estándar para este diagnóstico: el examen de electrolitos del sudor. El mismo evalúa, entre otros iones, los niveles de cloruro excretados. Una variedad de síntomas, incluyendo infecciones sinusales, disminución del crecimiento, diarrea e infertilidad, son el resultado de los efectos de la FQ sobre los distintos órganos.
Se trata de una de las enfermedades hereditarias fatales más comunes. Su prevalencia es mayor entre caucásicos; una de cada 25 personas de ascendencia europea es portadora asintomática de un gen para FQ, siendo la enfermedad genética más frecuente entre esta población. Los afectados pueden ser diagnosticados mediante pruebas genéticas prenatales; también por screnning neonatal o, durante la infancia temprana, por la mencionada prueba del sudor. No existe cura para la FQ, y la supervivencia media para estos pacientes se estima en 29 años, alcanzando valores más altos en algunos países (36,8 en EE.UU).[1] [2] En casos severos, el empeoramiento de la enfermedad puede imponer la necesidad de un trasplante de pulmón.
La FQ es causada por una mutación en un gen llamado regulador de la conductancia transmembrana de la fibrosis quística (CFTR, por sus siglas en inglés). Este gen interviene en la producción de sudor, jugos gástricos, y moco. Aunque la mayoría de las personas sanas tienen dos copias funcionales del gen, sólo una es necesaria para impedir el desarrollo de fibrosis quística. La FQ se desarrolla cuando ninguno de estos genes opera normalmente. En consecuencia, se la considera una enfermedad autosómica recesiva. El nombre fibrosis quística se refiere a los procesos característicos de cicatrización (fibrosis) y formación de quistes dentro del páncreas, reconocidos por primera vez en los años 1930.[3]
Síntomas y signos [editar]
La sintomatología de la fibrosis quística varía en función de la edad del individuo, el grado en que se ven afectados órganos específicos, la terapéutica instituida previamente, y los tipos de infecciones asociadas. Esta enfermedad compromete al organismo en su totalidad y muestra su impacto sobre el crecimiento, la función respiratoria, la digestión, y la reproducción. El periodo neonatal se caracteriza por un pobre aumento de peso y por obstrucción intestinal producida por heces densas y voluminosas. Otros síntomas aparecen, más tarde, durante la niñez y al inicio de la adultez. Éstos incluyen retardo del crecimiento, advenimiento de la enfermedad pulmonar, y dificultades crecientes por la malabsorción de vitaminas y nutrientes en el tracto gastrointestinal. Adicionalmente, el problema en la fertilidad se vuelve palpable una vez que se intenta conseguir la reproducción.
A la mayoría de los niños se les diagnostica fibrosis quística antes del primer año de vida, cuando la mucosidad pegajosa que afecta pulmones y páncreas, comienza a mostrar su impacto. En el tracto respiratorio, esas secreciones sirven como caldo de cultivo para diversas bacterias responsables de infecciones crónicas, con deterioro progresivo y permanente del parénquima pulmonar. Conforme se agrava la condición respiratoria, los pacientes sufren hipertensión pulmonar. Por otra parte, en el páncreas, el moco obstruye el tránsito de las enzimas sintetizadas por la glándula e impide que lleguen hasta los intestinos para digerir y absorber el alimento.
Enfermedad pulmonar y sinusal [editar]

Aspergillus fumigatus, un
hongo común que puede conducir al agravamiento de la enfermedad pulmonar en personas con FQ.
La enfermedad pulmonar resulta del bloqueo de las vías aéreas más pequeñas con el moco espeso característico de la fibrosis quística. La inflamación y la infección producen daño a los pulmones y cambios estructurales que conducen a una variedad de síntomas. En las etapas iniciales, comúnmente se presentan tos incesante, producción copiosa de flema, y una disminución en la capacidad aeróbica. Muchos de estos síntomas ocurren cuando ciertas bacterias (fundamentalmente, Pseudomonas aeruginosa) que normalmente viven en el moco espeso, crecen en forma descontrolada y causan neumonía. En estados avanzados de la FQ, los cambios en la arquitectura del pulmón producen dificultades respiratorias crónicas.
Otros síntomas incluyen expectoración de sangre o esputo sanguinolento, dilatación crónica de los bronquios o bronquiolos (bronquiectasia), elevación de la presión sanguínea en el pulmón, insuficiencia cardíaca, sensación de no estar recibiendo suficiente oxígeno o disnea, insuficiencia respiratoria y atelectasia; podría requerirse soporte ventilatorio.[4] Además de las infecciones bacterianas más comunes, las personas con FQ desarrollan con mayor facilidad otros tipos de enfermedades respiratorias. Entre éstas se encuentra la aspergilosis broncopulmonar alérgica, caracterizada por una respuesta de hipersensibilidad ante un hongo (moho) ordinario del género Aspergillus (Aspergillus fumigatus), que agudiza los problemas respiratorios. Otro ejemplo es la infección con el complejo Mycobacterium avium (MAC), grupo de actinobacterias emparentadas con Mycobacterium tuberculosis, que puede ocasionar daños mayores al pulmón, y que no responde a la terapéutica con antibióticos convencionales.
El moco en los senos paranasales es igualmente denso y pejagoso, y también puede causar oclusión de los orificios por donde los senos habitualmente drenan, lo cual hace que se acumulen secreciones que actúan como caldo de cultivo para los patógenos antes mencionados. En estos casos, se pueden presentar dolor facial, fiebre, secreción nasal profusa y cefaleas. En las personas con FQ, a menudo se observa crecimiento sobreabundante de tejido nasal (pólipos), a consecuencia de la inflamación por infección sinusal crónica. Estos pólipos pueden agravar la obstrucción de las vías respiratorias superiores e intensificar las dificultades respiratorias.[5] [6]
Enfermedad gastrointestinal, hepática y pancreática [editar]
Con anterioridad a la difusión de las pruebas prenatal y neonatal para fibrosis quística, era frecuente que la enfermedad se detectara al constatar que el recién nacido no podía expulsar sus primeras heces (meconio). El meconio puede obstruir completamente los intestinos y causar graves trastornos. Esta condición, llamada íleo meconial, ocurre en el 10% de los recién nacidos con FQ.[7] Asimismo, es también frecuente la asociación de FQ con protrusión de las membranas rectales internas (prolapso rectal), debida al mayor volumen fecal, a la malnutrición, y a la elevación de la presión intraabdominal por tos crónica.[8]
El moco glutinoso observado en el pulmón tiene su correlato en las secreciones espesas del páncreas, órgano responsable de proveer jugos digestivos que facilitan la descomposición química de los alimentos. Estas secreciones impiden el movimiento de las enzimas pancreáticas hacia el intestino y producen daño irreversible en el páncreas, a menudo acompañado de dolorosa inflamación (pancreatitis).[9] La deficiencia de enzimas digestivas se traduce en un impedimento para absorber los nutrientes, con la subsiguiente excreción de éstos en las heces: este trastorno es conocido como malabsorción. La malabsorción conduce a la desnutrición y al retardo en el crecimiento y desarrollo, ambos debidos a la baja biodisponibilidad calórica. Las personas con FQ tienen, en particular, problemas para absorber las vitaminas A, D, E, y K. Además de la afección pancreática, suelen experimentar acidez crónica, xerostomía, obstrucción intestinal por intususcepción, y constipación.[10] Los pacientes mayores desarrollan también el síndrome de obstrucción intestinal distal causado por las heces glutinosas.[11]
Estas secreciones también pueden causar problemas en el hígado. La bilis, producida por esta víscera para facilitar la digestión, podría bloquear las vías biliares, dañando los tejidos adyacentes. Con el tiempo, esta situación conduce a la cirrosis. Ese ese caso, resultan comprometidas funciones de primer orden, tales como las implicadas en la neutralización de toxinas, y en la síntesis de importantes proteínas (por ejemplo, los factores de coagulación, responsables de la coagulación sanguínea).[12]
Enfermedad endocrina y crecimiento [editar]
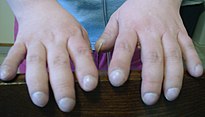
Las personas con FQ a menudo presentan malformación y agrandamiento de los dedos (dedos en palillo de tambor o hipocráticos)
El páncreas contiene los islotes de Langerhans, que son los responsables de producir insulina, una hormona que ayuda a regular los niveles de glucosa en sangre. Un daño en el páncreas puede provocar la pérdida de las células de los islotes y conducir a la diabetes.[13] Por otra parte, la vitamina D suplementada por la alimentación está implicada en la regulación del calcio y del fósforo. La baja disponibilidad de ésta, a causa de la malabsorción, conduce a la osteoporosis, aumentando el riesgo de sufrir fracturas.[14] Adicionalmente, las personas con FQ a menudo presentan, en manos y pies, una malformación denominada dedos en palillo de tambor, la cual se debe a los efectos de esta enfermedad crónica y a la hipoxia en sus huesos.
El retardo en el crecimiento es un sello distintivo de esta enfermedad. Los niños con FQ no logran, por lo general, ganar peso y altura en tasas comparables a las de sus pares; a menudo, sólo reciben diagnóstico apropiado una vez que se investigan las causas de este fenómeno. Las determinantes del retardo en el crecimiento son multifactoriales e incluyen la infección pulmonar crónica, la malabsorción de nutrientes en el tracto gastrointestinal, y el aumento de la demanda metabólica asociado a la afección crónica.
Infertilidad [editar]
Composición del esperma en afectados por FQ
| Valor normal | Fibrosis quística |
| pH | >8 | <7 |
| Ácido cítrico | 400-1.500 mg/100 ml | > 2.000 mg/100 ml |
| Fosfatasa ácida | 140-290 µg/ml | 760-1.140 µg/ml |
| Fructosa | 250-720 mg/100ml | 30-80 mg/100ml |
La infertilidad se manifiesta tanto entre los hombres como entre las mujeres con FQ. Al menos un 97 por ciento de los varones afectados son estériles.[15] Éstos producen esperma normalmente, pero carecen del vaso (conducto deferente), que conecta los testículos con los conductos eyaculadores del pene.[16] En muchos casos de ausencia congénita del conducto deferente se encontraron, durante la examinación por infertilidad, formas leves de FQ, que no habían sido diagnosticadas previamente.[17] Un veinte por ciento de las mujeres con FQ son estériles como consecuencia del moco cervical abundante y espeso, que interfiere con el paso de esperma. Por otro lado, en casos severos, la malnutrición produce alteraciones en la ovulación y causa amenorrea.[18]
Diagnóstico [editar]
La fibrosis quística puede diagnosticarse por tamizaje en recién nacidos, examen de electrolitos del sudor, o prueba genética. Al año 2006, en los Estados Unidos, el diez por ciento de los casos son detectados poco después del nacimiento como parte de los programas de pesquisa neonatal, que identifican niveles elevados en la enzima tripsina. Sin embargo, en la mayoría de los países estos exámenes no se realizan en forma rutinaria. Por esta causa, es frecuente que los afectados sólo reciban diagnóstico apropiado una vez que los síntomas fuerzan una evaluación para esta enfermedad. La prueba diagnóstica más comúnmente utilizada es el examen del sudor, descrito por Lewis E. Gibson y Robert E. Cooke en 1959,[19] usando electroforesis cuantitativa (iontoforesis) con un fármaco estimulante de la sudoración (pilocarpina). Esta sustancia, que posee carga positiva, se aplica sobre un electrodo positivo (+), en contacto con la piel. Luego, mediante el paso de corriente eléctrica, la droga migra por el tegumento hacia otro electrodo de carga opuesta (-), colocado a cierta distancia, hasta atravesar la epidermis, produciendo la estimulación de las glándulas sudoríparas y causando una sudoración controlada. Las muestras de sudor son luego colectadas en papel de filtro o en un tubo capilar y son analizadas, determinándose las concentraciones de sodio y cloruro. Las personas con FQ poseen niveles más altos de estos iones en el sudor. Una vez que el examen del sudor ha dado positivo, se realiza un diagnóstico más detallado y preciso, mediante la identificación de las mutaciones en el gen CFTR.[20]
Existen diversas pruebas para identificar eventuales complicaciones y controlar la evolución de la enfermedad. Las imágenes obtenidas por rayos X y TAC facilitan la detección de signos de lesión o infección en los pulmones. El cultivo de esputo, examinado por microscopio, provee información respecto de cuáles son las bacterias responsables, y permite escoger los antibióticos más efectivos. Las pruebas de función pulmonar miden las capacidades pulmonares, los volúmenes pulmonares y la rapidez con que éstos pueden ser movilizados (flujos aéreos). Por medio de tales exámenes, es posible determinar si es procedente un tratamiento con antibióticos o bien evaluar la respuesta al mismo. Los análisis de sangre pueden identificar problemas hepáticos, deficiencias vitamínicas, y revelar la irrupción de la diabetes. Los dispositivos DEXA o DXA (del inglés para "absorciometría de rayos X de energía dual"), se utilizan como prueba para determinar la presencia de osteoporosis. Por último, la cuantificación de elastasa fecal, facilita la detección de insuficiencia de enzimas digestivas.
Diagnóstico prenatal [editar]
Las parejas que están atravesando un embarazo o tienen planes respecto de la gestación, pueden ser evaluadas en busca de mutaciones del gen CFTR, con el objeto de determinar las probabilidades de que su hijo nazca con fibrosis quística. La prueba se suele realizar en uno de los padres o en ambos y, en caso de detectarse un riesgo elevado de FQ, se efectúa también en el feto. Debido a que el diagnóstico prenatal no habilita formas de tratamiento superiores o alternativas, la principal razón por la que se lleva a cabo es, en la práctica, proporcionar la posibilidad del aborto en caso de que el feto presente la enfermedad. La prueba para fibrosis quística en parejas se ofrece de manera generalizada en países como los Estados Unidos,[21] y el Colegio Americano de Obstetras y Ginecólogos (ACOG, por sus siglas en inglés) recomienda la prueba en parejas que poseen un historial de FQ entre sus familiares directos o parientes cercanos, así como también en aquellas con riesgo elevado debido a su filiación étnica.[22]
Debido a que el desarrollo de la FQ en el feto requiere que cada padre transmita una copia del gen CFTR mutante, y al alto costo del examen prenatal, la prueba suele realizarse, inicialmente, sólo en uno de los progenitores. Si éste resulta ser portador de una mutación del gen CFTR, entonces se examina al otro para determinar el riesgo de que su hijo tenga la enfermedad. La FQ puede resultar de más un millar de mutaciones diferentes y, al año 2006, no es posible efectuar estudios de laboratorio para cada una de ellas. La prueba se remite a analizar la sangre en busca de las más comunes, como ΔF508 — la mayoría de las modalidades disponibles comercialmente detectan no más de 32 variantes distintas. Si se conoce el dato de que una familia tiene una mutación poco común, esta última puede buscarse específicamente. Como consecuencia de que no todas las mutaciones conocidas son detectadas por las pruebas corrientes, un resultado negativo no garantiza que el niño vaya a estar libre de la enfermedad.[23] Por otro lado, dado que las mutaciones sondeadas son necesariamente aquellas más comunes en los grupos de más alto riesgo, las pruebas en etnias de bajo riesgo son menos exitosas, ya que las mutaciones más extendidas en estos grupos son menos frecuentes en la población general.
Las parejas en situación de riesgo, a menudo realizan pruebas adicionales durante el embarazo o antes de que éste se produzca. La fecundación in vitro con diagnóstico genético preimplantacional ofrece la posibilidad de examinar el embrión antes de su colocación en el útero. Esta prueba se realiza tres días después de la fecundación y procura determinar la presencia de genes CFTR anormales. Si, en un embrión, resultan identificados dos genes CTRF mutantes, éste será excluido de la transferencia, implantándose otro que cuente con, al menos, un gen normal.
Durante el transcurso del embarazo, es posible realizar pruebas tanto sobre la placenta (muestra de vellosidad coriónica) como sobre el líquido amniótico que rodea al feto (amniocentesis), con la ayuda del ultrasonido. Sin embargo, la biopsia de vellosidades coriónicas se correlaciona con riesgo de muerte fetal en una tasa de 1 en 100, y la amniocentesis, de 1 en 200,[24] por lo que es esencial determinar los beneficios adecuadamente para sopesar los riesgos, antes de proceder con la prueba. Alternativamente, algunas parejas eligen someterse a técnicas de reproducción asistida con óvulos donantes (recurriendo a la fecundación in vitro) o con esperma donante (inseminación artificial por donante).
Fisiopatología [editar]
La fibrosis quística ocurre cuando hay una mutación en el gen CFTR. La proteína creada por este gen se une a la membrana externa de las células en las glándulas sudoríparas, pulmón, páncreas, y otros órganos afectados. La proteína atraviesa esta membrana y actúa como un canal iónico conectando la parte interna de la célula (citoplasma) con el fluido extracelular. Este canal es mayormente responsable de controlar el paso de cloruro hacia (y desde) el medio interno. Cuando la proteína CFTR no funciona correctamente, este movimiento se ve restringido, reteniéndose cloruro en el espacio extracelular. Debido a que el cloruro tiene carga eléctrica negativa, los iones con carga positiva tampoco podrán cruzar la membrana citoplasmática, a causa de la atracción eléctrostática ejercida por los iones cloruro. El sodio es el más común entre los iones presentes fuera de la célula, y la combinación de sodio y cloruro da lugar al cloruro de sodio, el cual se pierde en grandes cantidades en el sudor de los individuos con FQ. Esta pérdida de sal constituye el argumento básico para explicar la eficacia del test del sudor.[4]
El mecanismo por el cual esta disfunción celular produce las manifestaciones clínicas antes descritas no se conoce con exactitud. Una de las teorías que intenta explicarlo, sugiere que la falla de la proteína CFTR para transportar el cloruro, determina la acumulación de abundante moco en los pulmones, creando un medio propicio (rico en nutrientes) para las bacterias, que logran así eludir al sistema inmunitario. También se postula que esta anomalía en la proteína CFTR induce un aumento paradójico en la captura de sodio y cloruro, lo que estimula la reabsorción de agua, y resulta en la formación de la mucosidad deshidratada y espesa. Otras teorías se enfocan en el fenómeno del movimiento de cloruro hacia el exterior de la célula, que también provoca desecamiento del moco y de las secreciones pancreáticas y biliares. En general, estas hipótesis coinciden en atribuir los mayores trastornos a la obstrucción de los conductos más delgados por las secreciones espesas y glutinosas en los distintos órganos afectados. Esta situación condiciona la infección crónica y promueve la remodelación estructural del pulmón, además de producir daño pancreático (mediado por las enzimas digestivas aglomeradas), y obstrucción de los intestinos por grandes bolos fecales.[4]
El papel de la infección crónica en la enfermedad pulmonar [editar]

Micrografía electrónica de barrido de la
bacteria Pseudomonas aeruginosa, asociada con frecuencia a las infecciones pulmonares graves que complican la FQ.
Los pulmones de las personas con fibrosis quística son colonizados e infectados por bacterias desde edades tempranas. Los microorganismos que se propagan en estos pacientes, prosperan en el moco anómalo acumulado en las vías respiratorias más estrechas. El moco glutinoso estimula el desarrollo de microambientes bacterianos (biofilms) que resultan difíciles de penetrar para las células inmunes y los antibióticos. Por su parte, los pulmones responden al daño continuo, infligido por las secreciones espesas y las infecciones crónicas, remodelando gradualmente las vías respiratorias inferiores (bronquiectasia), lo que vuelve a la infección aún más difícil de erradicar.[25]
Con el paso del tiempo, cambian tanto el tipo de bacterias que afectan a estos pacientes, como las características específicas con que las mismas se presentan. En una primera etapa, ciertas bacterias ordinarias como Staphylococcus aureus y Haemophilus influenzae colonizan e infectan los pulmones. Más tarde, sin embargo, prevalecen Pseudomonas aeruginosa (y, a veces, el complejo Burkholderia cepacia, integrado por diferentes especies de Burkholderia). Una vez diseminadas por las vías respiratorias, estas bacterias se adaptan al medio y desarrollan resistencia a los antibióticos convencionales. Pseudomonas puede adquirir ciertas características especiales, dando lugar a la formación de grandes colonias — estas cepas son conocidas como Pseudomonas "mucoide" y son raras en personas libres de la enfermedad.[25]
Uno de los modos en que la infección se propaga es por transmisión entre individuos con FQ.[26] En el pasado, era habitual que éstos participaran, en forma conjunta, de campamentos veraniegos y otras actividades de esparcimiento.[27] [28] Los hospitales alojaban a los pacientes con FQ en un área en común, y el equipamiento de rigor (por ejemplo, los nebulizadores)[29] no era esterilizado entre usos sucesivos.[30] Esto condujo a la transmisión de cepas bacterianas muy peligrosas entre grupos de pacientes. Actualmente, la rutina en establecimientos de atención sanitaria consiste en aislar a estos pacientes unos de otros; además, el personal a cargo de su cuidado, debe vestir batas y guantes para limitar la proliferación de cepas bacterianas virulentas.[31] Con frecuencia, los pacientes afectados por bacterias particularmente peligrosas reciben atención en días y en edificios diferentes a los asignados a quienes no tienen esas infecciones.
Se trata de una enfermedad autosómica recesiva que afecta a los pulmones, páncreas, hígado, glándulas sudoríparas, tubo digestivo y epidídimo. En su forma más común, una mutación de un aminoácido (falta una fenilalanina en la posición 508) conduce a una falla del transporte y localización en la membrana celular de la proteína CFTR.
El gen CFTR (acrónimo para el ingl. Cystic Fibrosis Transmembrane Regulator, "regulador de la conductancia transmembrana de la fibrosis quística") está localizado en el brazo largo del cromosoma 7, en la posición 7q31.2, ocupando 180.000 pares de bases: más precisamente, desde el par 116.907.252 al 117.095.950 del cromosoma. Es un gen de gran tamaño, que posee 250 kb y que incluye 27 exones.
Este gen codifica la síntesis de una proteína integrada por 1.480 aminoácidos, que transporta iones cloruro a través de las células epiteliales. En las personas con fibrosis quística, aquella proteína está ausente o bien se encuentra en proporciones sensiblemente menores a las habituales. Debido a que el agua sigue, por ósmosis, a los iones cloruro, la ausencia de cloruro por la falla en su transporte da como resultado una repleción de agua y la producción de moco viscoso.
Biología molecular [editar]
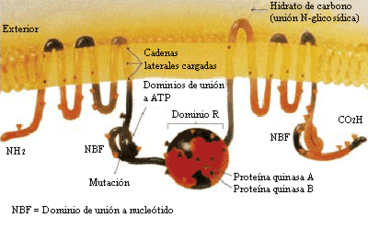
Proteína CFTR - estructura molecular de la proteína
La mutación más común, ΔF508, es una deleción (Δ) de tres nucleótidos que resulta en una pérdida del aminoácido fenilalanina (F) en la posición quingentésima octava (508) de la proteína. Esta mutación es responsable del setenta por ciento de los casos de FQ a nivel mundial y del noventa por ciento de éstos en los Estados Unidos. Sin embargo, existen más de 1.000 mutaciones distintas que pueden producir FQ. Son diversos los mecanismos por los cuales estas mutaciones causan problemas en la proteína CFTR. En particular, la mutación ΔF508, genera una proteína que no se pliega de manera normal y acaba siendo degradada por la célula. Varias mutaciones comunes en la población askenazí dan lugar a la síntesis de proteínas demasiado cortas, a causa de una conclusión anticipada de su producción. Otras mutaciones menos frecuentes originan proteínas que no utilizan la energía como es debido, no permiten que el cloruro cruce la membrana apropiadamente, o son degradadas a una tasa más rápida que la normal. Ciertas mutaciones pueden conducir también a una merma en la producción de copias de la proteína CFTR.[4]
Estructuralmente, el gen CFTR pertenece a la denominada superfamilia de transportadores ABC (acrónimo para el inglés ATP Binding Cassette, "casete de unión a ATP").[4] La estructura terciaria de la proteína codificada por este gen, consta de dos dominios capaces de hidrolizar adenosín trifosfato, lo que permite a la proteína utilizar energía en la forma de ATP. Asimismo, otro par de dominios, cada uno constituido por seis hélices alfa, posibilita el paso de la proteína a través de la membrana celular. La activación se concreta por reacción de fosforilación en un sitio de unión regulador, sobre todo mediante la proteína quinasa A (PKA, EC 2.7.11.11 —antes denominada cAPK o proteína quinasa dependiente del adenosín monofosfato cíclico).[4] El carboxilo terminal (C-) de la proteína está unido al citoesqueleto por interacción con dominios proteicos PDZ.[32]
Tratamiento [editar]

Cuidados respiratorios para la fibrosis quística con nebulizador y dispositivo para el control de secreciones
Un aspecto fundamental en la terapéutica de la fibrosis quística es el control y tratamiento del daño pulmonar causado por el moco espeso y por las infecciones, con el objeto de mejorar la calidad de vida del paciente. Para el tratamiento de las infecciones crónicas y agudas se administran antibióticos por vías intravenosa, inhalatoria y oral. También se utilizan dispositivos mecánicos y fármacos (en forma de inhaladores) para controlar las secreciones, y de esta manera descongestionar y desobstruir las vías respiratorias. Otros aspectos de la terapia se relacionan con el tratamiento de la diabetes con insulina, de la enfermedad pancreática con reemplazo enzimático, y de la infertilidad con técnicas reproductivas avanzadas. Adicionalmente, se postula la eficacia de distintos procedimientos, como el trasplante y la terapia génica, para resolver algunos de los efectos asociados a esta enfermedad.
Antibióticos para tratar la enfermedad pulmonar [editar]
Los antibióticos se prescriben siempre que exista sospecha de neumonía o se constate deterioro en la función pulmonar. Habitualmente, se los escoge en función del historial de infecciones que afectaron al paciente previamente. Muchas de las bacterias comunes en la fibrosis quística son resistentes a gran cantidad de antibióticos y requieren semanas de tratamiento intravenoso con vancomicina, tobramicina, meropenem, ciprofloxacina y piperacilina.
La terapia prolongada a menudo requiere hospitalización y canalización de una vía intravenosa permanente, como por ejemplo un catéter central insertado percutáneamente (PICC). Asimismo, es frecuente la indicación simultánea de antibióticos administrados por inhalación, como la tobramicina, la colistina y la gentamicina, por varios meses, con el objeto de mejorar la función pulmonar impidiendo la proliferación bacteriana.[33] [34] Algunos antibióticos orales como la ciprofloxacina o la azitromicina se utilizan a veces para ayudar a prevenir la infección o para controlarla una vez que está en curso.[35] En algunos casos pasan años entre sucesivas hospitalizaciones, mientras que en otros se requiere la internación cada año para poder realizar el tratamiento.
En tratamientos prolongados, varios de los antibióticos más comunes (como la tobramicina y la vancomicina) pueden causar pérdida de audición por ototoxicidad o problemas en los riñones. Con el objeto de prevenir tales efectos secundarios, es habitual medir cuantitativamente las concentraciones de estos medicamentos en sangre y, de ser necesario, ajustar la dosificación.
Otros métodos para tratar la enfermedad pulmonar [editar]
Son diversas las técnicas que se implementan con el objeto de fluidificar el esputo y facilitar su expectoración. En el medio hospitalario se utiliza la fisioterapia; un terapeuta practica una serie de maniobras mediante presiones y percusiones (palmoteo) ejercidas sobre el exterior del pecho (tórax) varias veces al día. Los dispositivos mecánicos que actúan bajo el mismo principio que aquellas técnicas básicas de drenaje postural, incluyen el ventilador de alta frecuencia oscilatoria y los aparatos de ventilación percusiva intrapulmonar, de los que existen modelos portátiles, adaptables al uso hogareño.[36] El ejercicio aeróbico es altamente beneficioso para las personas con fibrosis quística, ya que no sólo promueve la descongestión del esputo, sino que mejora la salud cardiovascular y el estado general.

Aparato de ventilación percusiva intrapulmonar
Entre las sustancias administradas por inhalación que ayudan a aligerar las secreciones y facilitan su expulsión, se encuentran la dornasa alfa y la solución salina hipertónica.[37] La dornasa es una desoxirribonucleasa (ADNasa o DNasa) humana recombinante, que descompone el ADN en el esputo, reduciendo así la viscosidad de este último.[38] La N-acetilcisteína (un derivado del aminoácido cisteína) también actúa fluidificando el esputo, pero las investigaciones y la experiencia disponibles han demostrado que los beneficios son poco significativos. Por último, broncodilatadores como el salbutamol y el salmeterol (ambos agentes, agonistas β2-adrenérgicos) o el bromuro de ipratropio (un antagonista del receptor colinérgico, derivado cuaternario de la atropina) se utilizan para aumentar el tamaño de las vías respiratorias más pequeñas, al relajar el músculo liso bronquial.
En la medida en que se agrava la condición pulmonar, puede requerirse soporte respiratorio mecánico. Por las noches, algunos pacientes deben usar máscaras especiales que actúan empujando el flujo aéreo hasta los pulmones. La ventilación no invasiva mediante máscara nasal y presión positiva (VPAP, por el acrónimo para el inglés variable positive airway pressure), ayuda a prevenir, durante el sueño, caídas significativas en los niveles sanguíneos de orxígeno. También puede usarse en el curso de la fisioterapia respiratoria para favorecer la expulsión de esputo.[39] Sin embargo, en casos severos, puede ser necesario implementar formas invasivas de asistencia respiratoria con intubación endotraqueal (esto es, colocación de un tubo o sonda en la tráquea).
Tratamiento de otros aspectos de la FQ [editar]

La
inyección intracitoplasmática de esperma (ICSI, por sus siglas en inglés) suele indicarse para revertir la infertilidad en hombres con FQ.
Los recién nacidos con íleo meconial típicamente requieren cirugía; por lo general, no sucede lo mismo en adultos con síndrome de obstrucción intestinal distal. El tratamiento de la insuficiencia pancreática basado en reemplazo de las enzimas digestivas menguadas permite que los intestinos absorban de manera apropiada nutrientes y vitaminas que, de otro modo, se perderían en la heces. Aun así, la mayoría de los individuos con FQ deben recibir dosis adicionales de vitaminas A, D, E y K a partir de suplementos, y seguir una dieta de alto valor calórico. La diabetes que suele acompañar la FQ se trata con inyecciones de insulina.[40] El desarrollo de osteoporosis puede prevenirse con la suplementación de vitamina D y calcio, y a menudo se trata con bifosfonatos.[41] En cuanto al retraso en el crecimiento, se procura contrarrestarlo mediante la inserción de un tubo de alimentación (gastrostomía) para aumentar así la ingesta de calorías a partir de nutrición adicional; también se administran con este fin inyecciones de hormona de crecimiento.[42]
Las infecciones de los senos paranasales suelen tratarse con un prolongado régimen de antibióticos. El desarrollo de pólipos, así como otros cambios estructurales de tipo patológico en el interior de los conductos nasales, pueden restringir el flujo aéreo y complicar el cuadro. Por este motivo, es frecuente la práctica quirúrgica en procura de aliviar la obstrucción y limitar el desarrollo de nuevas infecciones. También se administran corticosteroides intranasales, como la fluticasona, para reducir la inflamación.[43] Por otro lado, la infertilidad femenina puede combatirse recurriendo a técnicas de reproducción asistida. Aquella que afecta al hombre también tiene tratamiento: por ejemplo, mediante la inyección intracitoplasmática de esperma.[44]
Trasplante y terapia génica [editar]
Por lo general, se considera procedente el trasplante de pulmón en personas con deterioro progresivo de la función pulmonar y creciente intolerancia al ejercicio (fatiga o agotamiento muscular desproporcionados para el ejercicio realizado). Aunque el trasplante de un único pulmón es viable en otras enfermedades, en los pacientes con FQ ambos deben ser reemplazados ya que, de otro modo, las bacterias alojadas en el órgano remanente podrían infectar a aquél que ha sido trasplantado. Asimismo, puede practicarse simultáneamente un trasplante de páncreas o de hígado con el propósito de aliviar la enfermedad hepática o la diabetes.[45] La opción del trasplante de pulmón se evalúa cuando la función pulmonar se ve afectada en grado tal que se vea amenazada la supervivencia o se requiera la asistencia con dispositivos mecánicos.[46]
La terapia génica representa una vía promisoria en la lucha contra la enfermedad. Mediante esta técnica, se procura insertar una copia normal del gen CFTR en las células afectadas. Debido a la incapacidad de los retrovirus para alcanzar células que no se dividen, se han realizado análisis clínicos para insertar genes en adenovirus. En la actualidad, estos virus se están utilizando en ensayos en los que el gen CFTR normal se administra, por un método en aerosol, a las células epiteliales que revisten los pulmones (terapia génica in vivo). Se espera que los adenovirus inserten el gen normal, induciendo una función pertinente de los canales de cloro en estas células.
Algunos estudios han señalado que para prevenir las manifestaciones pulmonares de la fibrosis quística, sólo se requiere la expresión génica de entre un 5 y un 10% de los valores normales de proteína CFTR.[47] Un inconveniente de los adenovirus es que no se integran en el ADN de la célula huésped. Por lo tanto, finalmente se pierden, originando una expresión del gen transitoria y la necesidad de reintroducción del vector. Se han propuesto diversos abordajes y se han iniciado numerosos estudios clínicos pero, al año 2006, persisten múltiples obstáculos, que será preciso superar para que la terapia génica resulte exitosa.[48]
Epidemiología [editar]

Herencia mendeliana autosómica recesiva: dos mutaciones de
línea germinal (una de cada uno de los padres) para desarrollar la enfermedad; igualmente transmitida por hombres y mujeres.
La fibrosis quística es, entre las personas de ascendencia europea, la más frecuente de las enfermedades autosómicas recesivas potencialmente fatales. En los Estados Unidos, aproximadamente 30.000 individuos padecen FQ; en su mayoría, son diagnosticados a los seis meses de edad. Canadá tiene cerca de 3.000 habitantes con esta condición. Se estima que una de cada 25 personas de ascendencia europea y una de cada 29 personas de ascendencia askenazí son portadores de una mutación de fibrosis quística. Aunque es menos común en estos grupos, aproximadamente uno de cada 46 hispanoamericanos, uno de cada 65 africanos y uno de cada 90 asiáticos son portadores de al menos un gen CFTR anormal.[49] [50] [51] La Argentina representa una excepción en el contexto de América Latina, con una incidencia de casos mucho mayor a la media de la región y muy próxima a la registrada en Estados Unidos o Canadá, y una prevalencia de portadores sanos en la población general de 1 en 30.
La fibrosis quística se diagnostica tanto en hombres como en mujeres. Por razones que sólo en parte se conocen, la esperanza de vida al nacer resulta ser mayor entre los varones afectados que entre las mujeres.[52] Aquel indicador tiende a variar principalmente en función del alcance y la calidad de la atención suministrada por los sistemas de salud pública. En 1959, la supervivencia media en niños con FQ era de 6 meses. Para los nacidos en 2006 en los Estados Unidos, este valor treparía a los 36,8 años, de acuerdo a los datos compilados por la Fundación de la Fibrosis Quística.[2] La tasa de esperanza de vida ha evolucionado en forma análoga para buena parte de Occidente, exceptuando los países menos desarrollados, donde se reportan cifras sensiblemente menores, y en los cuales la mayoría de la población afectada no sobrevive más allá de los diez años de edad.
La Fundación de la Fibrosis Quística compila, además, información sobre el estilo de vida de los adultos estadounidenses con FQ. En 2004, la fundación reportó que el 91% de esta población había completado la enseñanza media, y el 54% había accedido a alguna forma de educación universitaria. Los datos en materia de empleo revelaron que el 12,6% de estos adultos estaba imposibilitado para trabajar (quedando fuera de la población económicamente activa), y el 9,9% estaba desocupado. Por otro lado, la información marital señaló que un 59% era soltero y un 36% estaba casado o viviendo en pareja. En 2004, 191 mujeres con FQ se encontraban embarazadas en los Estados Unidos.[53]
Teorías sobre la prevalencia de la FQ [editar]
Se estima que la mutación ΔF508 puede tener hasta unos 52.000 años de antigüedad.[54] Se han formulado numerosas hipótesis intentando explicar por qué una mutación letal como ésta ha persistido y se ha extendido entre la población humana. Algunas enfermedades autosómicas recesivas comunes como la anemia falciforme han revelado la propiedad de proteger a sus portadores de otras afecciones, concepto conocido como ventaja heterocigota. Con el descubrimiento de que la toxina del cólera requiere que sus húespedes sean proteínas CFTR normales para poder funcionar apropiadamente, se ha postulado que los portadores de genes CFTR mutantes obtuvieron el beneficio de la resistencia al cólera y a otras causas de diarrea.[55] Sin embargo, estudios posteriores no han confirmado esta hipótesis.[56] [57]
La presencia de proteínas CFTR normales es condición necesaria para el ingreso de Salmonella typhi (serotipo de Salmonella enterica, proteobacteria gram negativa del género Salmonella) en las células,[58] lo que sugiere que los portadores de genes CFTR mutantes podrían ser resistentes a la fiebre tifoidea. Sin embargo, ningún estudio in vivo ha confirmado esta hipótesis. En cualquiera de los casos, la baja incidencia de fibrosis quística fuera de Europa, en sitios donde tanto el cólera como la fiebre tifoidea son endémicos, carece de explicación inmediata.
Aunque el espectro clínico completo de la FQ no fue reconocido hasta los años 1930, ciertos aspectos fueron identificados mucho antes. Carl von Rokitansky describió un caso de muerte fetal con peritonitis meconial, una complicación del íleo meconial asociado con la fibrosis quística. El íleo meconial fue descrito por primera vez en 1905 por Karl Landsteiner.[59]
En 1938, Dorothy Andersen publicó un artículo intitulado Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study ("La fibrosis quística del páncreas y su relación con la enfermedad celíaca: un estudio clínico y patológico") en la revista American Journal of Diseases of Children. De esta manera, era la primera investigadora en definir esta entidad nosológica (denominada, por aquel entonces, "fibrosis quística del páncreas"), y en correlacionarla con los trastornos pulmonares e intestinales prominentes.[3] También postuló que era una enfermedad recesiva y utilizó el reemplazo de enzimas pancreáticas como tratamiento para los niños afectados. En 1952, Paul di Sant' Agnese descubrió anomalías en los electrolitos del sudor. Sobre la base de esa evidencia, se desarrolló y perfeccionó el examen del sudor durante el curso de la siguiente década.[60]
En 1985, investigadores de Londres, Toronto y Salt Lake City trazaron el mapa del gen CFTR en el cromosoma 7q. Cuatro años más tarde, en 1989, Francis Collins, Lap-Chee Tsui y John R. Riordan descubrieron la primera mutación para la FQ, ΔF508, en ese cromosoma. Investigaciones posteriores a aquel hallazgo, identificaron más de mil mutaciones diferentes que dan origen a la enfermedad. Lap-Chee Tsui lideró el equipo de científicos del Hospital for Sick Children (un hospital escuela en convenio con la Universidad de Toronto) que descubrió el gen responsable de la FQ. Se trata del primer trastorno genético dilucidado estrictamente mediante el proceso de genética inversa. Debido a que las mutaciones del gen CFTR son generalmente pequeñas, las técnicas de la genética clásica o formal no fueron capaces de determinar con precisión el gen mutante.[61] Utilizando marcadores proteicos, los estudios de ligamiento genético lograron trazar un mapa de la mutación del cromosoma 7. Las técnicas de paseo y salto cromosómicos sirvieron entonces para identificar y secuenciar el gen.[62]
La identificación de la mutación específica responsable de la FQ en un paciente puede ser útil para predecir la evolución de la enfermedad. Por ejemplo, los pacientes homocigotos para la mutación ΔF508 presentan, en casi todos los casos, insuficiencia pancreática y tienen, por lo general, un grado relativamente severo de afectación respiratoria. Sin embargo, existen excepciones que indican la posibilidad de que factores adicionales (quizás, genes en otros loci) intervengan en la expresión de la enfermedad. Por otro lado, la clonación del gen de la FQ ha abierto la posibilidad de la terapia génica, tal y como se ha descrito en la sección pertinente.
En la actualidad, los registros de la enfermedad indican que el 40% de los pacientes con fibrosis quística viven más allá de los 18 años.
Bibliografía [editar]
- Dapena Fernández, Francisco Javier. Fibrosis quística. Salobreña: Alhulia, 1998, 1ª ed. ISBN 8495136139
- Salcedo Posadas, Antonio; García Novo, María Dolores. Fibrosis quística. Madrid: Díaz de Santos, 1998, 1ª ed. ISBN 8479783680
- Segal, Edgardo. Fibrosis quística. Buenos Aires: Journal, 2004, 1ª ed. ISBN 9879773977
- Segal E, et al. "Consenso de Fibrosis Quística." Arch.argent.pediatr. 1999;97(3):188. Disponible en línea (PDF)
- ↑ Jorde, Lynn; Carey, John; White, Raymond. Genética médica. Madrid: Mosby, 1996. ISBN 8481741612
- ↑ a b New Statistics Show CF Patients Living Longer Cystic Fibrosis Foundation (26 de abril, 2006). Consultado el 26-07-2006.
- ↑ a b Andersen DH. "Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathological study." Am J Dis Child 1938; 56:344-399
- ↑ a b c d e f Rowe SM, Miller S, Sorscher EJ. "Cystic fibrosis." N Engl J Med. 2005 may 12;352(19):1992-2001. PMID 15888700
- ↑ Maldonado M, Martínez A, Alobid I, Mullol J. The antrochoanal polyp. Rhinology. 2004 dic;42(4):178-82. Rev. PMID 15626248
- ↑ Ramsey B, Richardson MA. Impact of sinusitis in cystic fibrosis. Allergy Clin Immunol. 1992 sep;90(3 Pt 2):547-52. PMID 1527348
- ↑ Eggermont E, De Boeck K. Small-intestinal abnormalities in cystic fibrosis patients. Eur J Pediatr. 1991 oct;150(12):824-8. Rev. PMID 1743211
- ↑ Kulczycki LL, Shwachman H. "Studies in cystic fibrosis of the pancreas; occurrence of rectal prolapse." N Engl J Med. 1958 ag 28;259(9):409-12. PMID 13578072
- ↑ Cohn JA, Friedman KJ, Noone PG, Knowles MR, Silverman LM, Jowell PS. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med. 1998 sep 3;339(10):653-8. PMID 9725922
- ↑ Malfroot A, Dab I. New insights on gastro-oesophageal reflux in cystic fibrosis by longitudinal follow up. Arch Dis Child. 1991 nov;66(11):1339-45. PMID 175564
- ↑ Khoshoo V, Udall JN Jr. Meconium ileus equivalent in children and adults. Am J Gastroenterol. 1994 feb;89(2):153-7. PMID 8304294
- ↑ Williams SG, Westaby D, Tanner MS, Mowat AP. Liver and biliary problems in cystic fibrosis. Br Med Bull. 1992 oct;48(4):877-92. PMID 1458306
- ↑ Moran A, Pyzdrowski KL, Weinreb J, Kahn BB, Smith SA, Adams KS, Seaquist ER. "Insulin sensitivity in cystic fibrosis." Diabetes. Agosto 1994;43(8):1020-6. PMID 8039595
- ↑ Haworth CS, Selby PL, Webb AK, Dodd ME, Musson H, McL Niven R, Economou G, Horrocks AW, Freemont AJ, Mawer EB, Adams JE. "Low bone mineral density in adults with cystic fibrosis." Thorax. 1999 nov;54(11):961-7. PMID 10525552
- ↑ McCallum TJ, Milunsky JM, Cunningham DL, Harris DH, Maher TA, Oates RD. "Fertility in men with cystic fibrosis: an update on current surgical practices and outcomes." Chest. 2000 oct;118(4):1059-62. PMID 11035677
- ↑ Dodge JA. "Male fertility in cystic fibrosis." Lancet. 1995 sep 2;346(8975):587-8. PMID 7650999
- ↑ Augarten A, Yahav Y, Kerem B, Halle D, Laufer J, Szeinberg A, Dor J, Mashiach S, Gazit E, Madgar I. "Congenital bilateral absence of vas deferens in the absence of cystic fibrosis." Lancet 344: 1473-74, 1994. PMID 7968122
- ↑ Gilljam M, Antoniou M, Shin J, Dupuis A, Corey M, Tullis DE. "Pregnancy in cystic fibrosis. Fetal and maternal outcome." Chest. 2000 jul;118(1):85-91. PMID 10893364
- ↑ Gibson LE, Cooke RE. "A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilising pilocarpine by iontophoresis." Pediatrics mar;23(3):545-9. PMID 13633369
- ↑ Stern, RC. "The diagnosis of cystic fibrosis." N Engl J Med 1997; 336:487. PMID 9017943
- ↑ ACOG Committee Opinion #325: "Update on Carrier Screening for Cystic Fibrosis". Obstet Gynecol 2005; 106:1465.
- ↑ American College of Obstetricians and Gynecologists and American College of Medical Genetics. Preconception and prenatal carrier screening for cystic fibrosis. Clinical and laboratory guidelines. American College of Obstetricians and Gynecologists, Washington, DC, octubre 2001.
- ↑ Elias, S, Annas, GJ, Simpson, JL. Carrier screening for cystic fibrosis: Implications for obstetric and gynecologic practice. Am J Obstet Gynecol 1991; 164:1077. PMID 2014829
- ↑ Tabor A, Philip J, Madsen M, Bang J, Obel EB, Norgaard-Pedersen B. Randomised controlled trial of genetic amniocentesis in 4606 low-risk women. Lancet. 1986 jun 7;1(8493):1287-93. PMID 2423826
- ↑ a b Saiman L. Microbiology of early CF lung disease. Paediatr Respir Rev. 2004;5 Supl A:S367-9. PMID 14980298
- ↑ Tummler B, Koopmann U, Grothues D, Weissbrodt H, Steinkamp G, von der Hardt H. Nosocomial acquisition of Pseudomonas aeruginosa by cystic fibrosis patients. J Clin Microbiol. 1991 jun;29(6):1265-7. PMID 1907611
- ↑ Centers for Disease Control and Prevention (CDC). Pseudomonas cepacia at summer camps for persons with cystic fibrosis. MMWR Morb Mortal Wkly Rep. 1993 jun 18;42(23):456-9. PMID 7684813
- ↑ Pegues DA, Carson LA, Tablan OC, FitzSimmons SC, Roman SB, Miller JM, Jarvis WR.Acquisition of Pseudomonas cepacia at summer camps for patients with cystic fibrosis. Summer Camp Study Group. J Pediatr. 1994 may;124(5 Pt 1):694-702. PMID 7513755
- ↑ Pankhurst CL, Philpott-Howard J. The environmental risk factors associated with medical and dental equipment in the transmission of Burkholderia (Pseudomonas) cepacia in cystic fibrosis patients. J Hosp Infect. 1996 Apr;32(4):249-55. PMID 8744509
- ↑ Jones AM, Govan JR, Doherty CJ, Dodd ME, Isalska BJ, Stanbridge TN, Webb AK. Identification of airborne dissemination of epidemic multiresistant strains of Pseudomonas aeruginosa at a CF centre during a cross infection outbreak. Thorax. 2003 jun;58(6):525-7. PMID 12775867
- ↑ Hoiby N. Isolation and treatment of cystic fibrosis patients with lung infections caused by Pseudomonas (Burkholderia) cepacia and multiresistant Pseudomonas aeruginosa. Neth J Med. 1995 jun;46(6):280-7. PMID 7643943
- ↑ Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998 jul 31;273(31):19797-801. PMID 9677412
- ↑ Pai VB, Nahata MC. Efficacy and safety of aerosolized tobramycin in cystic fibrosis. Pediatr Pulmonol. 2001 oct;32(4):314-27. Rev. PMID 11568993
- ↑ Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J Cyst Fibros. 2004 mar;3(1):23-8. PMID 15463883
- ↑ Hansen CR, Pressler T, Koch C, Hoiby N.Long-term azithromycin treatment of cystic fibrosis patients with chronic Pseudomonas aeruginosa infection; an observational cohort study. J Cyst Fibros. 2005 mar;4(1):35-40. PMID 15752679
- ↑ van der Schans C, Prasad A, Main E. Chest physiotherapy compared to no chest physiotherapy for cystic fibrosis. Cochrane Database Syst Rev. 2000;(2):CD001401. Rev. PMID 10796781
- ↑ Kuver R, Lee SP. Hypertonic saline for cystic fibrosis. N Engl J Med. 2006 abr 27;354(17):1848-51; respuesta del autor 1848-51. PMID 16642591
- ↑ Lieberman J. "Dornase aerosol effect on sputum viscosity in cases of cystic fibrosis." JAMA. 1968 jul 29;205(5):312-3. PMID 5694947
- ↑ Moran F, Bradley J. Non-invasive ventilation for cystic fibrosis. Cochrane Database Syst Rev. 2003;(2):CD002769. Rev. PMID 12804435
- ↑ Onady GM, Stolfi A. Insulin and oral agents for managing cystic fibrosis-related diabetes. Cochrane Database Syst Rev. 2005 jul 20;(3):CD004730. Rev. PMID 16034943
- ↑ Conway SP, Oldroyd B, Morton A, Truscott JG, Peckham DG. Effect of oral bisphosphonates on bone mineral density and body composition in adult patients with cystic fibrosis: a pilot study. Thorax. 2004 ag;59(8):699-703. PMID 15282392
- ↑ Hardin DS, Rice J, Ahn C, Ferkol T, Howenstine M, Spears S, Prestidge C, Seilheimer DK, Shepherd R. Growth hormone treatment enhances nutrition and growth in children with cystic fibrosis receiving enteral nutrition.J Pediatr. 2005 mar;146(3):324-8. PMID 15756212
- ↑ Marks SC, Kissner DG. Management of sinusitis in adult cystic fibrosis. Am J Rhinol. 1997 en-feb;11(1):11-4. PMID 9065342
- ↑ Phillipson GT, Petrucco OM, Matthews CD. Congenital bilateral absence of the vas deferens, cystic fibrosis mutation analysis and intracytoplasmic sperm injection. Hum Reprod. 2000 feb;15(2):431-5. PMID 10655317
- ↑ Simultaneous liver and pancreas transplantation in patients with cystic fibrosis. Transplant Proc. 2005 oct;37(8):3567-9. PMID 16298663
- ↑ Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, Yee JY, Kotloff RM, Lipson DA, Bunin GR. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med. 2006 mar 15;173(6):659-66. Epub 2005 dic 30. PMID 16387803
- ↑ Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol. 2002 nov;27(5):619-27. PMID 12397022
- ↑ Tate S, Elborn S. Progress towards gene therapy for cystic fibrosis.Expert Opin Drug Deliv. 2005 mar;2(2):269-80. Rev. PMID 16296753
- ↑ Rosenstein BJ and Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr. 1998 abr;132(4):589-95. Rev. PMID 9580754
- ↑ Hamosh A, Fitz-Simmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998 feb;132(2):255-9. PMID 9506637
- ↑ Kerem B, Chiba-Falek O, Kerem E. Cystic fibrosis in Jews: frequency and mutation distribution. Genet Test. 1997;1(1):35-9. Rev. PMID 10464623
- ↑ Rosenfeld, M, Davis, R, FitzSimmons, S, et al Gender gap in cystic fibrosis mortality. Am J Epidemiol 1997 145,794-803
- ↑ Cystic Fibrosis Foundation data (PDF) Consultado el 27/07/2006.
- ↑ Wiuf C. Do delta F508 heterozygotes have a selective advantage? Genet Res. 2001 ag;78(1):41-7. PMID 11556136
- ↑ Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 1994 oct 7;266(5182):107-9. PMID 7524148
- ↑ Cuthbert AW, Halstead J, Ratcliff R, Colledge WH, Evans MJ. The genetic advantage hypothesis in cystic fibrosis heterozygotes: a murine study. J Physiol. 1995 enero 15;482 (Pt 2):449-54. PMID 7714835
- ↑ Hogenauer C, Santa Ana CA, Porter JL, Millard M, Gelfand A, Rosenblatt RL, Prestidge CB, Fordtran JS. Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion. Am J Hum Genet. 2000 dic;67(6):1422-7. Epub 2000 oct 30. PMID 11055897.
- ↑ Pier GB, Grout M, Zaidi T, Meluleni G, Mueschenborn SS, Banting G, Ratcliff R, Evans MJ, Colledge WH. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature. 1998 may 7;393(6680):79-82. PMID 9590693
- ↑ Busch R. "On the history of cystic fibrosis." Acta Univ Carol [Med] (Praga). 1990;36(1-4):13-5. PMID 2130674
- ↑ Di Sant' Agnese PA, Darling RC, Perera GA, et al. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: clinical implications and relationship to the disease. Pediatrics 1953; 12:549-563.
- ↑ Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989 sep 8;245(4922):1066-73. Erratum in: Science 1989 sep 29;245(4925):1437. PMID 2475911
- ↑ Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989 sep 8;245(4922):1059-65. PMID 2772657
Véase también [editar]
Enlaces externos [editar]
- En inglés
- Advertencia: Wikipedia no es un consultorio médico.
- Si cree que requiere ayuda, por favor consulte con un profesional de la salud.


 claro está, también, Feliz Navidad.
claro está, también, Feliz Navidad. claro está, también, Feliz Navidad.
claro está, también, Feliz Navidad.
















